Chemical Reactivity Visualised
A 10-minute read

Conferences are valuable sources of inspiration for scientists. Latest results are presented, celebrated, or challenged. Poster sessions, coffee breaks, and conference dinners create a vibrant atmosphere, where novel hypotheses are formed, and in the best cases, seed the idea for important, new developments. But inspiration is not enough, as we all know. Method development is tedious and requires a favourable mixture of expertise, perseverance, and frustration tolerance.
When Richard Henderson suggested refining scattering factors for electron diffraction at the Murnau Conference in 2016, he did not know that he had just talked to the right person. Tim Grüne not only had profound knowledge of the SHELX software package, thanks to more than a decade spent in the group of the late George Sheldrick. Equally important, he had started to explore the potential of electron diffraction. With remarkable success. In 2018, he demonstrated that electron diffraction could be successfully deployed for the rapid structure determination of microcrystalline molecular compounds [1]. The publication in Angewandte Chemie was among those recognised by Science as a runner-up to the Breakthrough of the Year [2]. More recently, he was part of an international collaboration that successfully elucidated the polar habit and chiral structure of the malaria pigment hemozoin [3]. The exact crystal structure and that of the crystal surfaces have been the subject of intensive research for more than two decades. This is because preventing the growth of hemozoin crystals in the vacuoles of Plasmodium is an important strategy in the development of active substances against malaria.
Almost a decade after the Murnau Conference, Tim Grüne and his PhD student Soheil Mahmoudi, in collaboration with scientists from Vienna, Tennessee, and Switzerland, published a novel method to refine electron diffraction structures. It allows for the first time partial charges of chemical compounds to be derived using a generally applicable experimental method. In their publication in Nature [4], the authors demonstrate that their method of ionic scattering factors (iSFAC) modelling allows for the partial charges of all atoms of a chemical compound to be refined. This goes beyond the capabilities of X-ray diffraction, X-ray spectroscopy, EELS, NMR, vibrational spectroscopy, etc., which produce observables that can be combined with quantum chemical calculations to assign partial charges.
Why does it work? Because electrons interact with the electrostatic potential, which is very sensitive to changes in the charge distribution, as shown by earlier work of Koji Yonekura. He and co-workers calculated the ionic scattering factors of atoms that compose biological molecules. The electron scattering factors strongly depend on the ionic state of the atom, in particular at low scattering vectors s, as illustrated in Figure 2. This is in distinct contrast to X-ray scattering factors, which hardly vary between neutral and charged atoms [5]. Parameterisation of the calculated electron scattering factors forms the basis of the novel refinement, because it allowed Tim Grüne to adopt the well-established program SHELXL for the refinement, as described in detail by the authors in the Methods section. The iSFAC modelling method extends the classical crystal structure refinement by one parameter. This parameter is the fraction of ionic scattering that contributes to the electron scattering of each atom.
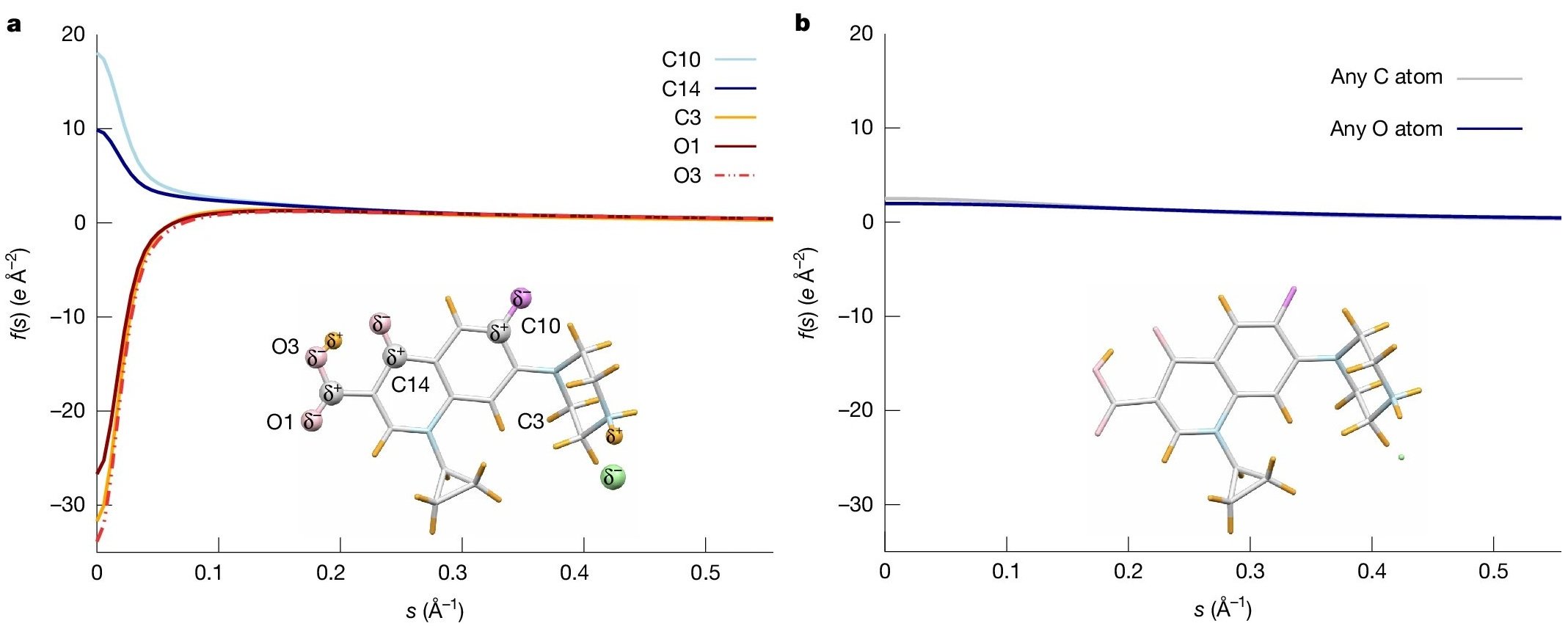
Why does it matter? Because partial atomic charges give rise to electrostatic forces that control molecular structure, interactions, and reactivity. Changes in the charge distribution in molecules alter bond length and induce chemical reactions, including the breaking and formation of chemical bonds.
Accurately determining partial charges has far-reaching impact across chemical synthesis, materials science, and theoretical chemistry, and is essential for parameterizing molecular-dynamics simulations—the discipline’s “computational microscope” for chemical processes. It will foster our understanding of biological function and how materials behave. In medicine, they influence the therapeutic effect and potential side effects of a drug by affecting the absorption, distribution, and metabolism of the active molecule.
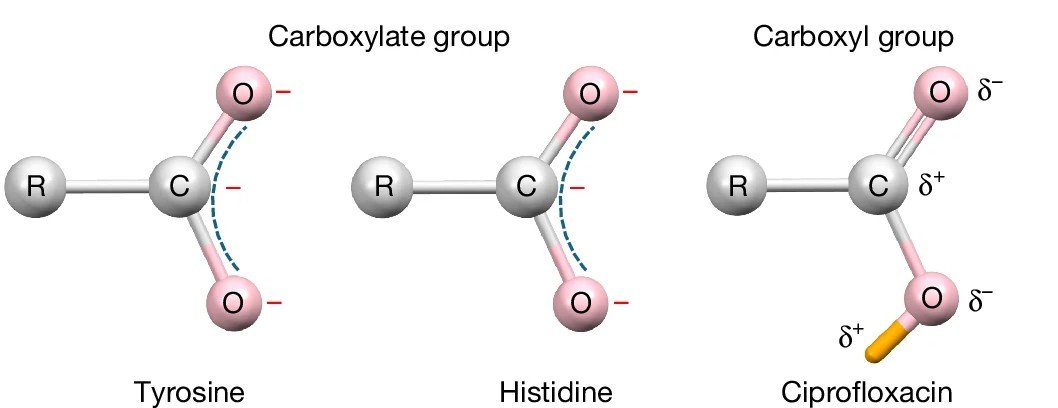
Why read the paper? To learn about the remarkable simplicity, sensitivity, and robustness of the method. Simplicity: Traditional electron crystallography workflows can be applied, and no specialised software is required. The use of a detector with high dynamic range, such as DECTRIS SINGLA, is advantageous for collecting complete and accurate data for both strong and weak reflections to the highest resolution. Sensitivity: While X-ray-based methods struggle to resolve hydrogen atoms, iSFAC permits the full refinement of their coordinates, atomic displacement parameters (ADPs), and partial charges. It also enhances the hydrogen signal compared to conventional refinement. Not surprisingly, the use of iSFAC refinement yields better agreement factors compared to standard kinematic refinement. The importance of hydrogen is illustrated in the different partial charge patterns of carboxylate and carboxylic acid groups, as shown in Figure 3. While the carbonyl carbon in the deprotonated COO− groups has a negative partial charge, it carries a positive partial charge in the carboxylic acid.
Robustness: The authors demonstrate that the modelling method yields very consistent results up to 0.95 Å resolution, and can still be used with small deviations up to 1.2 Å. The Pearson correlation coefficient of the partial charges refined from data collected from a zeolite at room and at cryogenic temperatures is as high as 0.91. Finally, important for electron diffraction methods, the modelling is independent of crystal thickness and dynamical effects.
This latest breakthrough marks a further milestone in the remarkable development of electron crystallography and microscopy during the last decade. Meanwhile, the next frontier appears clear: Richard Henderson speculates that it might become possible to observe the charged state of amino acid side chains in proteins in single particle analysis [6], and some groups are already exploiting the potential [7].
References:
- Tim Gruene et al., Angew. Chem. Int. Ed. 2018, 57, 16313-16317.
- https://www.science.org/content/article/breakthrough-2018#rapid-structure.
- Paul Klar et al., ACS Cent. Sci. 2024, 10, 1504-1514.
- Soheil Mahmoudi et al., Nature 2025, 645, 88-94.
- Koji Yonekura et al., IUCrJ 2018, 5, 348-353.
- Ardan Patwardhan et al., Curr. Opin. Struct. Biol. 2025, 92, 10300.
- Thomas Bick et al., BBA Adv. 2024, 5, 100113.




